



Het model achter waterstofbruggen en soortgelijke complexen is fundamenteel onjuist, tonen Amsterdamse onderzoekers aan in Chemistry – A European Journal.
De meeste chemici kennen de waterstofbrug als een vrij sterke intermoleculaire interactie tussen een waterstofatoom en bijvoorbeeld zuurstof, stikstof of halogenen (denk aan de basenparen in DNA). Het idee hierachter zou dan zijn dat waterstof elektropositief is en dus een relatief positieve lading heeft. Het andere molecuul zou elektronegatieve atomen bevatten en dus een relatief negatieve lading hebben. Dit veroorzaakt een elektrostatische aantrekking, zoals bij magneten.
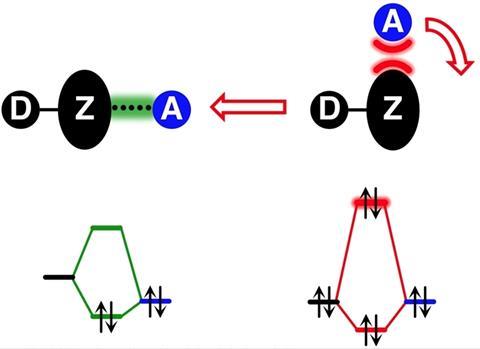
’Er ontstonden echter problemen toen men zich realiseerde dat twee elektronegatieve atomen elkaar óók konden aantrekken’, zegt Lucas de Azevedo Santos, postdoc bij de TheoCheM-groep van de Vrije Universiteit Amsterdam. Je vindt dit fenomeen in zogenaamde stikstofgroep-, zuurstofgroep- en halogeenverbindingen in de vorm van Lewiszuur-base interacties tussen respectievelijk groep 15, 16 en 17 elementen (in het Engels pnictogen, chalcogen en halogen genoemd). ’Om dit te verklaren is het σ- (sigma) hole model gepostuleerd. Dit model beschouwt het binding donerende Lewiszuur als een positief gebied op het oppervlak rond het molecuul, terwijl de binding accepterende Lewisbase wordt behandeld als een geconcentreerde negatieve puntlading.’ Maar uit nieuwe berekeningen met moleculaire orbitaaltheorie blijkt dat dit model twee zeer belangrijke aspecten verwaarloost: de fysica achter zowel de elektrostatische interacties als de directionaliteit van de intermoleculaire interacties.
’Let wel, het σ-gat bestaat wel degelijk, want het is experimenteel gedetecteerd’, zegt De Azevedo Santos. “Maar het punt is dat er geen causaal verband bestaat tussen het σ-gat en het bestaan en de vorming van intermoleculaire complexen.’ Om hun punt te bewijzen deden De Azevedo Santos en zijn collega’s kwantumchemische berekeningen met FmZ∙∙∙F–, waarbij m = 1-3 en Z een stikstofgroep-, zuurstofgroep- of halogeenatoom is.
Als het σ-hole model klopt, zou je verwachten dat de sterkte en de richting van de binding worden bepaald door de aantrekkingskracht van het positieve gebied en de negatieve puntlading, maar dat is niet het geval. Dit model kan de fysica die eruit zou moeten volgen niet verklaren. Zoals de auteurs in het artikel schrijven: ‘De reden voor dit gedrag is eenvoudig: moleculen zijn geen elektrostatische potentialen op oppervlakken en atomen zijn geen puntladingen!’ De Azevedo Santos: ’Moleculen bestaan uit atomen met een positieve kern en een negatief geladen elektronendichtheid eromheen. Het σ-gat model behandelt de moleculen fysisch gewoon verkeerd.’
Maar de auteurs hadden niet alleen kritiek op het model, ze poneerden in het artikel ook een eigen verklaring. ’Wij stellen voor dat de basis voor deze interacties “intermoleculaire covalente interacties” zijn. Al deze bindingen en complexen delen elektronen!’ legt de postdoc uit. ‘Technisch gesproken bestaan deze bindingen uit een lege orbitaal – de σ* LUMO als elektronenacceptor – en een gevulde orbitaal – de HOMO. Deze orbitalen overlappen elkaar in de stikstofgroep-, zuurstofgroep- en halogeenverbindingen, waardoor binding ontstaat.’
Maar waarom is dit belangrijk? ‘Omdat kennis van de fundamentele fysica helpt de natuur beter te begrijpen’, zegt De Azevedo Santos. ’Als je de interacties begrijpt, kun je deze kennis gebruiken voor het rationele ontwerp van moleculen. Er zijn bijvoorbeeld biologische en synthetische systemen met de functie om anionen over lipidemembranen te transporteren. Deze systemen gebruiken die specifieke interacties. Dus als je weet hoe ze werken, kun je de activiteit specifiek voor deze transporter verbeteren.’
De Azevedo Santos, L. et al. (2023) Chem. Eur. J. e202203791, DOI: 10.1002/chem.202203791.






Nog geen opmerkingen