




Simulaties bevestigen dat het gebruik van elektronen-stuwende groepen op radicalen de resulterende C‒C-binding zwakker maakt. Ook de reden van dit zogenoemde captodatieve effect is nu ontdekt, staat in een paper uit ChemistryEurope.
Radicalen, moleculen met een ongepaard elektron, staan te boek als zeer reactieve stoffen. Maar als je je substituenten goed kiest, dan kun je die reactiviteit redelijk afzwakken. Gebruik je zowel elektron-stuwende als elektron-zuigende groepen, dan spreek je over het captodatieve effect. Dát het werkt, is bij organici bekend, maar hóe het werkt is een ander verhaal. Eva Blokker, Matthias Bickelhaupt en collega’s van de TheoCheM-groep aan de Vrije Universiteit Amsterdam gebruikten orbitaaltheorie en simulaties om het mechanisme achter het captodatieve effect in kaart te brengen. Hun resultaten staan in het nieuwe open access tijdschrift ChemistryEurope.
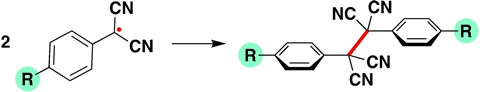
In de paper keken ze naar para-gesubstitueerde fenyldicyanomethylradicalen, waarbij ze varieerden met het substituent op de fenylgroep. Zet je daar waterstof of cyaan (zuigende groep) neer, dan is de C‒C-binding tussen de twee radicalen sterker dan wanneer je er -OMe of -NMe2 (beide stuwende groepen) aan hangt, die de binding juist zwakker maken.
Schild
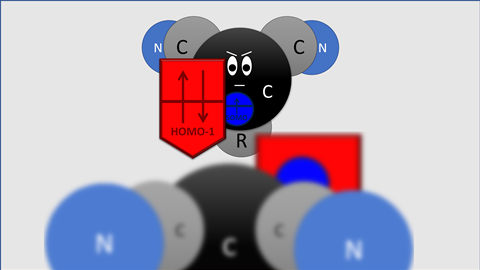
Blokker en co vonden dat de elektronen die door -OMe of -NMe2 gestuwd worden op het koolstofatoom naast het radicaal gaan zitten. ‘Op die plek zitten de elektronen in een soort π-orbitaal dat overlapt met het orbitaal waar het koolstofradicaal in zit’, legt Matthias Bickelhaupt, hoogleraar theoretische chemie aan de VU, uit. ‘Dat overlap-orbitaal is groter dan het singly occupied molecular orbital van het elektron [SOMO, red.], en vormt als het ware een “schild” om het SOMO heen, vandaar onze term lone-pair-shielded radical.’
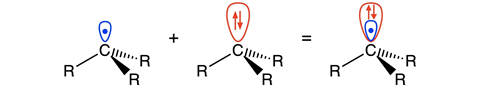
Het pseudo-vrije-elektronpaar zit in het zogenoemde highest occupied molecular orbital - 1 (HOMO-1) van het benzyllische koolstof. Die is dus zo groot dat het de C‒C-binding tussen twee radicalen sterk verzwakt door Pauli-repulsie. ‘De oude verklaring is fysisch gezien verkeerd’, zegt Bickelhaupt. ‘De oude verklaring kijkt naar de verhoging of verlaging in energie van de radicaalorbitalen van de twee betrokken gesubstitueerde methylradicalen. Maar dat fenomeen heeft principieel geen effect op de sterkte van de elektronenpaarbinding en verklaart dus niet de verzwakking van de C–C binding. Onze simulaties laten nu voor het eerst het mechanisme zien, dus dat een lager liggend gevuld orbitaal door stuwende groepen groter in amplitude wordt, zo over het orbitaal van het elektron heen valt en de binding dus actief tegenwerkt.’
Fundamenteel vs. experimenteel
De sleutelvraag is of dit effect meer voorkomt dan alleen in dit molecuul. ‘Ik vermoed’, zegt Bickelhaupt voorzichtig, ‘dat dit fenomeen heel breed voorkomt. Wat je nodig hebt is een lokaal π-symmetrisch orbitaal op een atoom dat naast een koolstofradicaal zit. Zulke substituenten die andere elektronen kunnen huisvesten bestaan er genoeg: halogenen, aromatische structuren, pseudo-halogenen. Dus ik kan me voorstellen dat dit daar dan ook optreedt. Ik hoop dat collega-onderzoekers hier ook op duiken.’
Met het begrip van deze zeer fundamentele kennis, kun je toch ook heel praktische kanten op. ‘Nu we weten waarom dit soort radicalen zich zo gedragen, kun je veel gerichter synthese doen: eerst nadenken over goede kandidaten, dan computationeel structuren verkennen en dan uiteindelijk experimenteel de laatste stap zetten.’
Blokker, E. et al. (2023) ChemistryEurope e202300006, DOI: 10.1002/ceur.202300006









Nog geen opmerkingen