
Simulations confirm that the use of electron withdrawing groups on radicals weakens the resulting C–C bond. The reason for this so-called captodative effect has now been discovered, according to a paper published in ChemistryEurope.
Radicals, molecules with one unpaired electron, are known to be highly reactive substances. But if you choose your substituents well, you can reduce this reactivity to some extent. If you use both electron donating and electron withdrawing groups, this is called the captodative effect. That it works is well known in organic chemistry, but how it works is another question. Eva Blokker, Matthias Bickelhaupt and colleagues from the TheoCheM group at VU Amsterdam have used orbital theory and simulations to map the mechanism behind the captodative effect. Their results are published in the new open access journal ChemistryEurope.
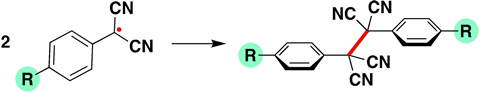
In the paper, they looked at para-substituted phenyl dicyanomethyl radicals, while varying the substituent on the phenyl group. If you put hydrogen or cyan (withdrawing group) at the para position, the C–C bond between the two radicals is stronger than if you use -OMe or -NMe2 (both donating groups), which actually make the bond weaker.
Shield
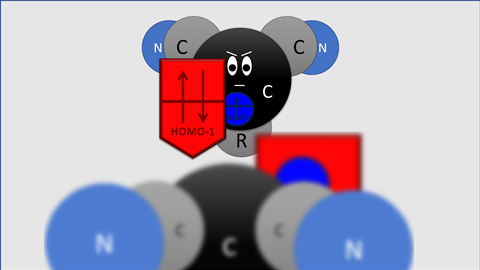
Blokker and co found that the electrons donated by -OMe or -NMe2 sit on the carbon atom next to the radical. ‘There, the electrons sit in a kind of π-orbital that overlaps the orbital containing the carbon radical,’ explains Matthias Bickelhaupt, professor of theoretical chemistry at VU. ‘This overlapping orbital is larger than the singly occupied molecular orbital of the electron [SOMO, ed.] and forms a “shield” around the SOMO, so to speak, hence our term lone-pair shielded radical.’
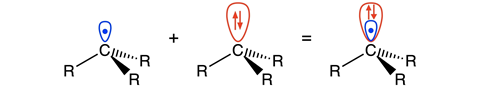
Image: Blokker, E. et al. (2023) ChemistryEurope e202300006
The pseudo-free electron pair is located in the so-called highest occupied molecular orbital - 1 (HOMO-1) of the benzyl carbon. It is so large that it strongly weakens the C-C bond between two radicals due to Pauli repulsion. ’The old explanation is physically wrong’, says Bickelhaupt. ’It is based on the increase or decrease in the energy of the radical orbitals of the two substituted methyl radicals involved. But this phenomenon has no effect on the strength of the electron-pair bond and therefore does not explain the weakening of the C-C bond. Our simulations now show for the first time the mechanism: A lower-lying filled orbital becomes larger in amplitude due to donating groups, falls over the orbital of the electron and thus actively opposes the bond.’
Fundamental versus experimental
The key question is whether this effect occurs in more than just this molecule. ‘I suspect’, says Bickelhaupt cautiously, ’that this phenomenon is very widespread. What you need is a local π-symmetric orbital on an atom next to a carbon radical. There are many such substituents that can host other electrons: halogens, aromatic structures, pseudohalogens. So I can imagine that this could happen there too. I hope that other researchers will also look into this.’
But understanding this very fundamental knowledge can also lead in very practical directions. ’Now that we know why these kinds of radicals behave the way they do, you can do a much more targeted synthesis: first think of good candidates, then explore structures computationally, and finally do the final step experimentally.’
Blokker, E. et al. (2023) ChemistryEurope e202300006, DOI: 10.1002/ceur.202300006






Nog geen opmerkingen