
The strength of hydrogen bonds between two molecules is also influenced by the other atoms in the molecule, shows Spanish-Dutch research in Physical Chemistry Chemical Physics.
The hydrogen bond plays an important role in the interaction between biomolecules such as DNA and proteins, where it contributes to self-assembly. If you want to exploit this principle yourself, you need to understand hydrogen bonding well. That’s where recent research by Celine Nieuwland, Célia Fonseca Guerra and colleagues from VU Amsterdam and the University of Barcelona comes in, which looks at the influence of the molecular backbone of hydrogen-bonding molecules.
Normally, scientists in general only look at the interactions that take place ‘in the foreground’, i.e. between the partially electropositive hydrogen atoms (H-O and H-N) and the electronegative atoms such as oxygen and nitrogen. The fact that the prevailing model of hydrogen bonding is incorrect has been discussed before. Building on this, the researchers wanted this work to further challenge the idea that you can explain hydrogen bonding strength using only the so-called frontier (H-N/O) atoms.
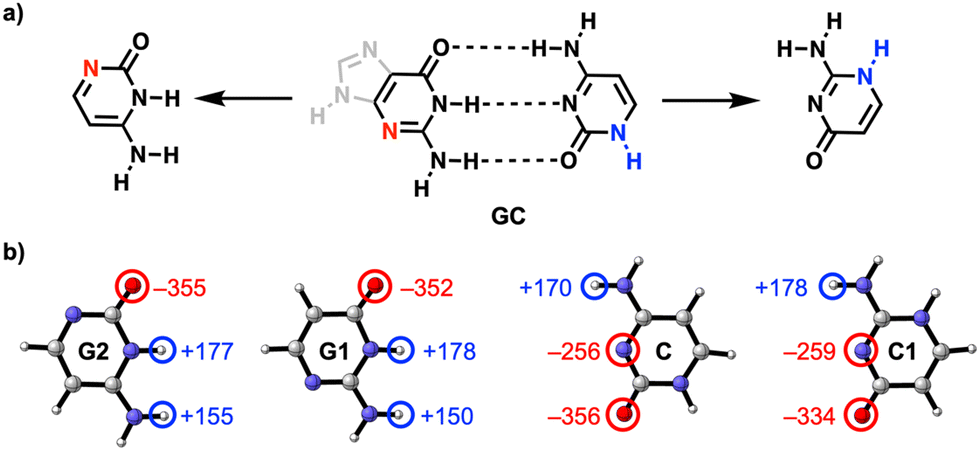
They did this by calculating the hydrogen bonds between the relevant parts of guanine (G) and cytosine (C), two molecules that are also found in DNA, where they form a complementary base pair. Both molecules have a nitrogen atom in the ring that is not directly involved in the hydrogen bonds.
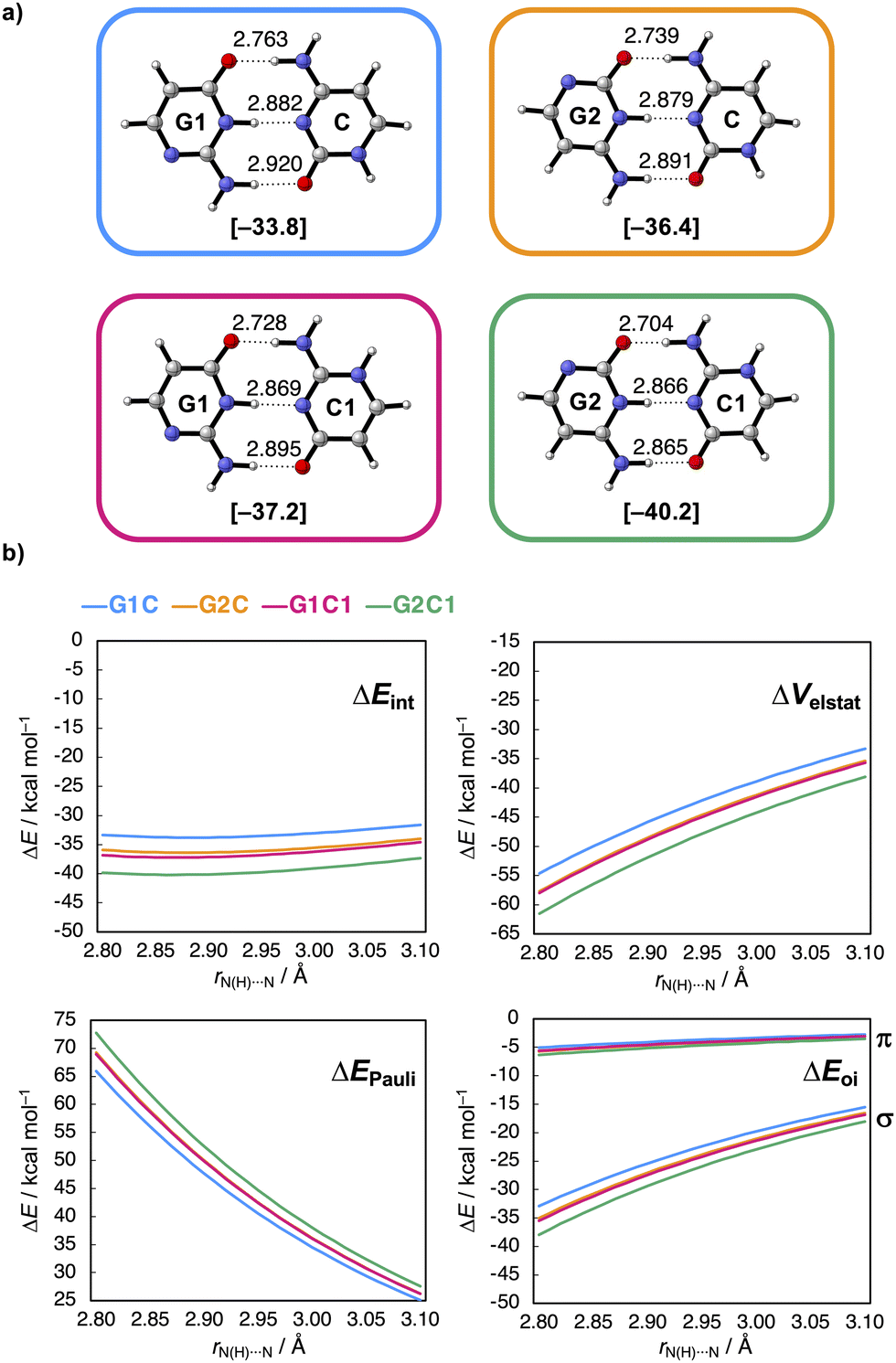
The researchers’ calculations showed that if you move this nitrogen and NH group up two places in the ring, so that they end up closer to the C=O and NH2 groups respectively, the hydrogen bonds become stronger (see figure): they measured a rather large difference in interaction energy of 6.5 kcal/mol. This is because charge accumulates in the molecules after the shift, which in turn leads to stronger electrostatic as well as σ-orbital interactions between the molecules.
This supports two observations: first, the electrostatic interactions seem to depend not only on the frontier atoms, but also on atoms further away. Second, the molecular orbitals are delocalised throughout the molecule, so their energies are affected when you adjust the backbone of the molecule. With this concept, you can take a smarter approach to designing biomolecules and hydrogen bonding systems.
Nieuwland, C. et al. (2024) PCCP 26, DOI: 10.1039/D3CP05244C






Nog geen opmerkingen