
DFT calculations can be used to find out why chiral-at-metal complexes lose their chirality and how this can be prevented, reports Chemistry A European Journal. ‘Knowing how it works gives you the opportunity to do something with it.’
Almost all biomolecules (from sugars to amino acids) are chiral, and many drugs work because they act on chirality. But to make chiral molecules, you also need chiral molecules or catalysts. ‘In metal catalysis, you use metal complexes to which a chiral ligand is attached’, says Koop Lammertsma, professor emeritus at the Vrije Universiteit Amsterdam and the University of Johannesburg. ‘But this is rather strange: the metal in such a complex is often already chiral, so it is somewhat redundant to add a chiral substituent. In addition, such ligands are often expensive or difficult to synthesise.’
Racemisation
But there is a reason why researchers are doing it, says Lammertsma. ‘Complexes in which the metal itself is chiral (chiral-at-metal) are not stable. Complexes with five or six substituents or ligands can twist and turn. Hexa-coordinated complexes in particular turn into their mirror-image isomer. If you start with one mirror-image isomer, but some of it turns into the other mirror-image isomer, this is called racemisation.’ Together with George Dhimba and Alfred Muller from the University of Johannesburg, Lammertsma investigated what causes racemisation and what can be done to stop it. ‘With this paper, we wanted to understand more about how these processes work. Knowing how it works gives you the opportunity to do something with it.’
Twists
If you have six loose ligands around a metal, then – depending on how big they are – they’ve got far too much freedom to move. So in order to study these twists and turns, you have to restrict the freedom a little bit, and you do that by using chelate ligands. A chelate is a molecule that can coordinate in two places on the metal, so you restrict the freedom of movement quite a bit. Lammertsma and colleagues used this observation in combination with DFT calculations to determine the different twists and how much energy they require.
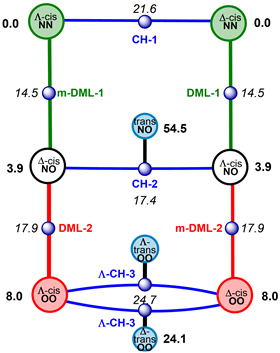
They simulated a hexa-coordinated molybdenum complex with two asymmetric chelates and two double-bonded oxygen atoms [(MoO2(acnac)2, acnac = β-ketoiminate]. The complex can spin in a number of ways, namely with Bailar, Ray-Dutt, Conte-Hippler or Dhimba-Muller-Lammertsma twists. The latter two were recently discovered by the three researchers and appear to play a crucial role in racemisation. In fact, they are the twists that require the least amount of energy to move from one mirror-image isomer to the other. For the sake of clarity, they have summarised all the different racemisation pathways in a graph.
Boost
What are the practical implications? With these calculations you can find out how a complex goes from one enantiomer to the other. If you know this, you can adjust your ligands to prevent these flips, so that your complex remains stable in the right enantiomer, the researchers also show in their paper. Lammertsma: ‘This can give a boost to chiral-at-metal catalysis, I’m absolutely sure of it. These theoretical calculations make it possible to see structurally how racemisation works in your complex. This will allow us to move away from complex, expensive and environmentally unfriendly ligands.’
Lammertsma hopes that this work will be widely disseminated so that others can benefit from it. ‘It has been nine years since my retirement that we published this paper, and yet it is the best paper of my career. It has a good chance of having more impact than all my previous papers if it gets picked up.’
Dhimba, G., Muller. A en Lammertsma, K. (2023) Chem Eur J e202302516, DOI: 10.1002/chem.202302516






Nog geen opmerkingen