

A paradigm in organic chemistry that has been in use since 1931 has turned out to be wrong, Amsterdam scientists found. It is not carbon s-p hybridisation, but steric repulsion that causes the variation of e.g. C-H bond lengths. ‘This discovery has been in the making for about twenty years.’
It is actually part of the standard rules in (physical) organic chemistry textbooks: the bond length between two atoms becomes shorter if you increase the s-character of one of the two atoms. So the distance between atoms decreases in the hybridisation trend sp3-sp2-sp, like the C-H bond in ethane, ethene and ethyne. ‘Before our discovery, it was actually never investigated whether this phenomenon really works that way with quantitative quantum chemical methods,’ says Matthias Bickelhaupt, professor of theoretical chemistry at Free University (VU) Amsterdam. ‘Yes, the hybridisation rule seems plausible and you can often observe corresponding correlations. But whether s-p hybridisation is the reason for bond length trends has never been proven quantum mechanically.’
Nevertheless, Bickelhaupt had already suspected this through research carried out some twenty years ago. ‘At that time we investigated why the nitrogen-fluorine bond in the molecule N2F+ is the shortest bond ever measured’, he continues. ‘Compared to NF4+, the N-F bond in N2F+ was shorter and we first thought that it was due to hybridisation. But after more research, it turned out not to be the case.’
‘Rules are useful, but as a scientist you just want to know how things really are’
Although the hybridisation trend was present, it was not responsible for the short bond. Bickelhaupt: ‘What we then found out was that the N-F bond of NF4+ experienced much more steric repulsion due to the larger number of substituents compared to N2F+.’ In the conclusion, he and his fellow researchers therefore raised the question of whether this steric effect could also be seen in the much more common C-H and C-C bonds.
Unfortunately, all sorts of things came in between, but a few years ago a team consisting of Bickelhaupt, Pascal Vermeeren, Willem-Jan van Zeist, Trevor Hamlin and Célia Fonseca Guerra took up the subject again. And indeed, the same steric effect can also be seen in those ubiquitous bonds. It resulted in a cover position in Chemistry - A European Journal (see also the illustration).
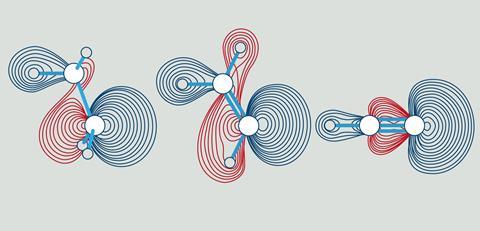
Qualitative and quantitative
The researchers used a quantitative molecular orbital theory (MO) in combination with an energy decomposition analysis. This forms a model that both reveals the mechanisms and causal relationships and quantifies the associated effects. ‘This enables us to determine both qualitatively and quantitatively how important certain effects are.’
So what are the details concerning steric repulsion? The C-H bond overlap does indeed reach its optimum at shorter distances when you go from sp3 to sp. Interestingly, however, that optimum lies at very short distances, roughly at about 0.7 Å and thus far below the C-H equilibrium distances of 1.07 to 1.10 Å. Around that final bond length, a totally different mechanism dominates, namely the steric repulsion (Pauli-repulsion) between the substituents (H-, CH3-, H2C= or HC≡) around the carbon atom of the C-H bond in question. This steric repulsion is a manifestation of the so-called Pauli repulsion between electrons of equal spin,’ Bickelhaupt explains, ‘and it obviously decreases when you go from 4, to 3 to 2 substituents around the carbon atoms in ethane, ethene and ethyne respectively.’
‘You just can’t solve a lot of things by blindly trying them out’
The most important implication of this discovery, in Bickelhaupt’s opinion, is the replacement of the ancient empirical rules. ‘Those rules are useful, don’t get me wrong, but as a scientist you just want to know how things really work, whether there is a real connection. The hybridisation model turns out not to be like that. With our quantitative MO theory, we know more and more about the real causal relationships and can therefore design molecules more specifically. This saves a lot of computational and experimental trial and error, and therefore time and money. And let’s face it: a lot of things you won’t ever solve by blindly trying them out.’
Polymerisation
Despite this being a very fundamental subject, there are also practical applications. ‘We have a collaboration with Nouryon, in which we use this kind of insight to design polymerisation initiators, for example,’ Bickelhaupt explains. ‘I can’t say too much about it, but the fundamental knowledge really helps in the design process.’
It remains to be seen when this new paradigm will be found in textbooks. Bickelhaupt knows, for instance, that this kind of research can take ten to twenty years to find its way into mainstream classes. Bickelhaupt: ‘I do suspect that this discovery will penetrate a little faster, perhaps with the next edition of a book on organic chemistry. Eventually the new knowledge will diffuse into the lecture halls.’
Overhauling paradigms
The paradigm shift from hybridisation to steric repulsion concerning bond lengths is not the only thing the Amsterdam theoretical chemistry group is working on. Thomas Hansen, postdoc in Bickelhaupt’s group, is looking at Lewis acid catalysts, for example. ‘Catalysis is very important in chemistry, but to apply it effectively you need to understand it well’, says Hansen. ‘If a Lewis acid catalyses a Diels-Alder reaction, it was assumed that the LUMO (lowest unoccupied molecular orbital) would be lowered, which would result in a stronger orbital change effect. The LUMO lowering does occur, but the sum of orbital interactions does not become stronger. The actual reason that the catalyst accelerates the reaction is that it lowers the electron density on the dienophile, resulting in a reduced steric hindrance.’ Bickelhaupt adds: ‘In technical terms, there is less closed-shell repulsion because the occupied pi-orbitals of the dienophile have a smaller amplitude. This research is also just starting to catch on in the community.’





Nog geen opmerkingen